Corrosion of Carbon Steels
Corrosion of Carbon Steels
Carbon (C) steel is the most widely used engineering material. Despite its relatively limited corrosion resistance, C steels are used in large tonnages in marine applications, power plants (both nuclear based power and fossil fuel based power), metal processing equipment, power transmission, transportation, chemical processing, petroleum production and refining, pipelines, mining, and construction etc.
The annual cost of metallic corrosion to the total economy of a country is very high. Because C steels represent the largest single class of alloys in use, both in terms of tonnage and total cost, it is easy to understand that the corrosion of C steels is a problem of enormous practical importance. This, of course, is the reason for the existence of entire industries devoted to providing protective systems for iron and steel. Because of corrosion, the design aspects are also very important. Indeed, design changes are often the most efficient manner of dealing with a particular corrosion problem.
C steels also often called mild steels have limited alloy content, usually less than 2 % of the total of all the additions. These levels of additions do not generally produce any remarkable changes in general corrosion behaviour. One possible exception to this statement is the weathering steels, in which small additions of copper (Cu), chromium (Cr), nickel (Ni), and/or phosphorus (P) produce significant reductions in corrosion rate in certain environments. At the levels present in the C steels, the usual impurities have no significant effect on corrosion rate in the atmosphere, neutral waters, or soils. Only in the case of acid attack is an effect observed. In this latter case, the presence of P and sulphur (S) markedly increase the rate of attack. Indeed, in acid systems, the pure irons appear to show the best resistance to attack.
In solving a particular corrosion problem, a dramatic change in attack rate can often be attained by altering the corrosive environment. The deaeration of water and the addition of corrosion inhibitors are two examples that have broad application in the area of aqueous corrosion. These and other techniques are useful in some cases.
Since corrosion being a multifaceted phenomenon, it is generally helpful to attempt to categorize the various types of corrosions. This is usually done on an environmental basis. The different categories on this basis are atmospheric corrosion, aqueous corrosion, and some other corrosion types of interest, such as corrosion in soils, concrete, and boiler plants.
Atmospheric corrosion
Atmospheres are often classified as being rural, industrial, or marine in nature. Such a classification is of course a gross oversimplification of the situation. It is possible to find places along the seacoast that have heavy industrial pollution in the atmosphere. Such places have both marine and industrial atmospheres. Moreover, two distinct rural environments can differ widely in average annual temperature and rainfall and can therefore have considerably different corrosive tendencies.
Industrial expansion in former rural areas can easily change the aggressiveness of a particular place. Also, long term trends in the environment, such as changes in rainfall patterns, mean temperature, and perhaps acid rain, can make extrapolations from past behaviour less reliable. Other factors that limit the usefulness of available atmospheric exposure data are the general nonlinearity of weight loss due to corrosion with time and the fact that most atmospheric-corrosion data are presented as an average over the entire test panel surface. Most atmospheric exposure data for C steels show a decrease in the rate of attack with time of exposure so that extrapolations of such data to times longer than those covered by the exposure data can lead to an overdesign in cross section. Also, in many cases, the average weight loss per unit area is of less concern than the time to perforation. This factor is more related to localized attack, which can be concealed by the averaging of data, as is done in weight loss determinations.
Given all the experiences mentioned above, a design engineer is well justified in using atmospheric-corrosion data as more of indicative nature than quantitative. The difference in corrosion rate is usually greater than any effect which can be produced by small changes in composition of the steel. This underscores the fact that in dealing with the corrosion of C steels the modification of design or environmental factors is normally more effective than changing the grade of steel.
While examining the corrosion rate of C steels in different environments, it can be seen that the marine environments tend to be more aggressive and that cold environments are generally less aggressive than warm places. The average annual temperature cannot, in general, be isolated from the moisture effect, because most of the more tropical exposure places are also located in areas with high humidity.
Corrosion film formation and breakdown
The corrosion of C steel in the atmosphere and in many aqueous environments is best understood from a film formation and breakdown standpoint. It is an unavoidable fact that iron (Fe) in the presence of oxygen (O2) and/or water is thermodynamically unstable with respect to its oxides. Thus, there is no doubt that the C steel is going to be corroded, but the rate at which it is going to be corroded is important. In the absence of film formation and with a constant environment, one can expect the oxidation rate to be constant. Conversely, if the corrosion product film that forms isolates the steel from the corrosive environment, then a zero corrosion rate can be expected after the initial film formation period. A tightly adherent film that permits only diffusion transfer of the reactants is normally characterized by a corrosion rate that decreases with the square root of the exposure time.
Because these idealizations are rarely encountered in the corrosion of C steels, it is obvious that other issues that tend to disrupt stable film formation are operative. These issues can be external, such as erosion by wind or rain, or they may be internal to the film itself, such as stresses caused by the different specific volumes of the steel and its oxide. It is the complexity of these various breakdown processes that makes a quantitative prediction of corrosion of the C steels very difficult, makes extrapolations of data from short term behaviour to longer times so challenging, and makes precise predictions of behaviour at a particular place based on observations at different places virtually impossible.
The corrosion of iron in the atmosphere proceeds by the formation of hydrated oxides. The half-cell reactions can be expressed as follows.
1/2O2 + H2O + 2e = 2(OH)- (cathodic)
Fe =Fe+ + 2e (anodic)
Further the following reactions can then occur.
Fe2+ + 2(OH)- = Fe(OH)2
2Fe(OH)2 + H2O + 1/2O2 = 2Fe(OH)3
The hydrated oxides can lose water during dry periods and revert to the anhydrous ferrous and ferric oxides. In addition, a layer of magnetite (Fe3O4 or FeO·Fe2O3) often forms between iron oxide (FeO) and hematite (Fe2O3). Actually, the various oxides and hydroxides of Fe form a rather complicated system of compounds. The compound FeOOH has been found to exist in three different crystal forms plus an amorphous form. The occurrence of the various oxide types is dependent on pH, availability of O2, various atmospheric pollutants, and the composition of the steel, as in weathering steels containing Cu and P. The actual nature of the corrosion film is important because FeO and FeOOH seem to be more adherent than Fe3O4 and Fe2O3, and therefore more likely to slow the corrosive attack, but the higher oxides and oxy-hydroxides are more prone to spalling action.
Atmospheric factors in atmospheric corrosion
Since there is a substantial variation in the corrosion rates of C steels at different atmospheric locations, it is only logical to know which of the factors contribute to these differences. Although the prediction of corrosivity is still not possible, it appears that humidity, temperature, and the levels of chloride, sulphate, and probably other atmospheric pollutants present each exert an influence on the corrosion rate of C steels.
Humidity and atmospheric pollutants
Since atmospheric corrosion is an electrolytic process, the presence of an electrolyte is required. This does not mean that the steel surface is to be awash with water, but what is needed is a very thin absorbed film of water. During an actual exposure, the steel spends some portion of the time awash with water because of rain or splashing and a portion of the time covered with a thin adsorbed water film. The portion of time spent covered with the thin water film depends quite strongly on the relative humidity at the place of exposure Fig 1 (i). This fact has encouraged many corrosion engineers and scientists to investigate the influence of the time of wetness on the corrosion rate. These studies have shown that time of wetness, although an important factor cannot be considered in isolation when estimating corrosion rates. A good example of this fact is demonstrated in Fig 1 (ii), in which the weight gain of Fe is plotted as a function of relative humidity for an exposure of 55 days in an environment containing 0.01 % sulphur dioxide (SO2). In the lower right hand corner of Fig 1 (ii) is the measured corrosion rate for Fe exposed for the same time in an SO2 free environment at 99 % relative humidity. The increase in corrosion rate produced by the addition of SO2 to the environment is substantial, as shown in Fig 1 (iii), which shows actual exposure data from four different exposure places with varying levels of S pollution in the atmosphere. The accelerating effect of S is noticeable.
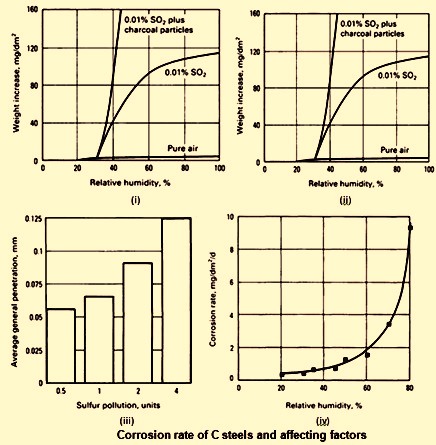
Fig 1 Corrosion rate of C steels and affecting factors
Another area of interest is the apparent existence of a critical humidity level below which the corrosion rate is small. As shown in Fig 1(ii), the critical humidity in an SO2 containing atmosphere is around 60 %. This behaviour contrasts with that of C steel in contact with particles of sea salt, as shown in Fig 1(iv). In Fig 1(iv), the corrosion rate shows a steady increase with increasing humidity.
Although there is a scarcity of data, it seems reasonable that oxides of nitrogen (NOx) in the atmosphere also show an accelerating effect on the corrosion of C steel. Indeed, any gaseous atmospheric constituent capable of strong electrolytic activity is to be suspected as being capable of increasing the corrosion rate of C steel. Similarly temperatures also have the accelerating effect on corrosion rate. Also it is noticed that there is an accelerating effect of chloride ions (Cl-) on the atmospheric corrosion.
Effects of alloying additions
Since C steels are by definition not very highly alloyed, it is not surprising that most grades of C steels do not show large differences in atmospheric corrosion rate. However, alloying can make changes in the atmospheric corrosion rate of C steels. The elements generally found to be most beneficial in this regard are Cu, P, Ni, Cr, and silicon (Si). Of these, the most prominent example is that of Cu. Increases from 0.01 % to 0.05 % have been shown to decrease the corrosion rate by a factor of two to three. Additions of the above elements in combination are generally more effective than when they are added alone. However, such additions do not have additive effects. The effectiveness of these elements in retarding corrosion also appears to depend on the corrosive atmosphere, with the most benefit appearing in industrial atmospheres.
Kinetics of atmospheric corrosion
The rate of atmospheric corrosion of C steels is not constant with time but usually decreases as the length of exposure increases. This fact indicates the difficulty in using most of the available atmospheric corrosion data in any quantitative form. Most of the available data consists of weight loss due to corrosion averaged over the time of exposure, which is often variable. Such corrosion rate calculations are misleading, especially when the exposure time is short, because the ensuing rate of attack can be considerably lower. Several studies have shown that the amount of corrosion occurring as a function of time can be expressed as per the equation ‘W= (Kt)n’ (to the power n) where ‘W’ is the weight loss of steel due to corrosion, ‘t’ is the exposure time in years, and ‘K’ and ‘n’ are empirical constants. The goodness of fit seems to be excellent, but until more data from various atmospheric exposure places are analyzed, this equation is of limited value. Since the values of ‘K’ and ‘n’ depend on the alloy system and the exposure place, a great deal of work is to be done before general use of the above equation can be made in real applications. The one possible exception is when data for the exposure situation and alloy system are already known. In this case, the above equation can be very useful in estimating long term corrosion behaviour from as little as 2 years of data, though 3 to 4 year data give a better estimate.
Maybe the most important thing which the above equation points is that it is impossible to describe either the extent or rate of corrosion under atmospheric conditions with a single parameter, which is what much of the reported corrosion data persists in doing.
When the results of a several year exposure experiment are condensed to a single value, such as the average loss per year or the total loss for the exposure period, then it is not possible to estimate the values of the kinetic parameters governing the system. Without the values of these parameters, extrapolation of results to longer exposure periods becomes quite unreliable. When good estimates for the kinetic parameters are available, extrapolations to 7 or 8 year performance from 1 and 2 year data have been found to agree within 5 % of observed performance.
Soil corrosion
The response of C steels to soil corrosion depends primarily on the nature of the soil and certain other environmental factors, such as the availability of moisture and O2. These factors can lead to extreme variations in the rate of attack. For example, under the worst conditions, a buried container may perforate in less than 1 year, although archaeological excavations in arid desert regions have uncovered iron tools dating back hundreds of years. Because of this intrinsic variability, any cumulative weight loss data or kinetic information is virtually worthless for design use. Some general rules can be formulated, however. Soils with high moisture content, high electrical conductivity, high acidity, and high dissolved salts are normally more corrosive. At the other extremes of character, the soil is virtually inert to C steel. The effect of aeration on soils is somewhat different from the effect of aeration on water because poorly aerated conditions in water can lead to accelerated attack by sulphate reducing anaerobic bacteria.
The effect of low levels of alloying additions on the soil corrosion of C steels is modest at best, with most common additions showing no significant benefit. Some data seem to show a small benefit of 1 % Cu + 2.5 % Ni over plain C steel. However, it is debatable whether this improved performance justifies the cost of alloying additions. This decision must involve other factors, such as the cost of a failure analysis and the cost of replacement.
Interestingly, it has been demonstrated that both the weight loss and maximum pit depth in soil corrosion can be represented by an equation of the form ‘Z = (at)m’ (to the power m) where ‘Z’ is either the weight loss of maximum pit depth, ‘t’ is time of exposure, and ‘a’ and ‘m’ are constants that depend on the specific soil corrosion situation. This equation is similar to the equation for the kinetics of the atmospheric corrosion. The similarities between these two equations may be due to the general applicability of a power law relationship, but because both types of corrosion are fundamentally electrolytic in nature, there may be some underlying connection.
Corrosion in concrete
The corrosion process in concrete is such that it tends to create conditions that increase the rate of attack. This phenomenon is related to the fact that the various corrosion products of C steels have a larger specific volume than the steel itself. The increase in volume of the corrosion products causes stresses that can lead to cracks in the concrete. These cracks allow easier access for the attacking medium and therefore more rapid attack. When the cracks are open to the exterior, corrosion products can be washed out and often lead to cosmetically objectionable stains. Of greater concern is the case of pre-stressed concrete. In this material, the corrosion process can lead to loss of structural strength and eventual failure. Of the cases studied, the failures of pre-stressed structures are not associated with stress corrosion, but are merely due to the loss of load-bearing area of steel.
The presence of chloride ions appears to be the principal cause of steel corrosion in concrete. Various attempts at reducing or eliminating the corrosion problem have focused on protective coatings for the steel members (galvanizing, polymer coating, painting etc.), decreasing the concrete permeability, increasing the depth of concrete cover, or eliminating the chloride ion through the use of sealants etc. Although many of these approaches have shown some degree of success, the application of cathodic protection has been the most successful in arresting corrosion. .
There does not appear to be any significant body of data relating the severity of corrosion to the composition of the steel reinforcing member. It is likely that alloying can reduce the overall rate attack, but whether the reduction is significant because of the added cost is problematic.
Carbon steel for boiler service
Corrosion in boilers is a special case of aqueous corrosion that involves high temperatures. The availability of O2 appears to be the rate determining step insofar as general attack is concerned. In closed loop systems, the initial O2 supply of the water is rapidly consumed in the early stages of film formation so that corrosion rates are usually not a problem. In open loop systems, deaeration is usually adequate for eliminating general corrosion problems.
Of more concern in boiler systems is the occurrence of pitting. In pitting corrosion, both dissolved O2 and carbon dioxide (CO2) promote attack. Deaeration is useful in stopping the O2 attack, but CO2 pitting is more effectively handled by maintaining an alkaline pH in the water. Surface deposits of corrosion products, mill scale, or even oil films have occasionally been implicated in the pitting attack of boilers.
C steel boilers are also susceptible to a form of stress corrosion cracking (SCC) termed caustic cracking. This can occur even when the bulk concentration of caustic is below the danger level because of a variety of processes that can lead to localized accumulations of caustic well in excess of average concentrations. Because alkalinity is desired to avoid pitting attack, it is obvious that procedures for preserving the benefit of pit suppression without incurring caustic cracking are necessary. Fortunately, such procedures are available.
Liquid metal corrosion
Liquid metals can attack plain C steels in many ways. First, if Fe is soluble in the liquid metal, dissolution occurs until the liquid metal is saturated. At this point, dissolution ceases in an isothermal system. Unfortunately, real systems almost always have temperature gradients. The presence of these gradients leads not only to dissolution in the high temperature portions of the systems but also to a precipitation or plating out in the colder areas. This precipitation decreases the solute content below the level that was present at the higher temperature, thus enabling additional dissolution to occur at the high temperature areas. The end result of this material transfer process is that there is either the complete dissolution of the container on the high-temperature side of the system or, in the case in which the liquid metal is contained in channels or pipes, a blockage on the cold side from the continued precipitation.
A second mode of attack involves the formation of intermetallic alloys of the steel with the liquid metal. If the particular intermetallic is poorly adherent or if it can be spalled off by mechanical or thermal stresses (this exposes fresh surface to form more alloy), perforation can eventually result. On the other hand, if the intermetallic alloy that forms is tightly adherent and resistant to cracking, it can act as a barrier to diffusion and can slow the rate of attack.
Another form of attack that can occur and which is more injurious is the inter-granular penetration and/or dissolution by liquid metal. The unfortunate aspect of this mode of attack is that it can result in a loss of strength without any large weight loss or change in appearance. In this respect, it resembles the more familiar aqueous inter-granular corrosion.
Also, there is the phenomenon of liquid metal embrittlement, which needs the presence of both stress and a liquid metal. The cracks that occur during the embrittlement process are mostly inter-granular or trans-granular. The process seems to be similar in many ways to SCC. C steels have been reported to undergo embrittlement by lithium, indium, cadmium, zinc, tellurium, and various lead-tin solders.
Resistance of plain C steels to liquid metal corrosion is given below.
- Sodium and sodium-potassium alloys – Plain C and low alloy steels are generally suitable for long term use in these media at temperatures up to 450 deg C. Beyond these temperatures, stainless steels are usually needed.
- Lithium – Lithium is somewhat more aggressive to plain C steels than the sodium or sodium-potassium. As a result, low alloy steels are not to be considered for long term use above 300 deg C. At higher temperatures, the ferritic stainless steels show better results.
- Cadmium – With cadmium, low alloy steels show good serviceability up to 700 deg C.
- Zinc – Most of engineering metals and alloys show poor resistance to molten zinc, and C steels are no exception.
- Antimony – Low C steels have poor resistance to attack by antimony.
- Mercury – Although plain C steels are virtually not attacked by mercury under non-flowing or isothermal conditions, the presence of either a temperature gradient or liquid flow can lead to drastic attack. The corrosion mechanism seems to be one of dissolution, with the rate of attack increasing rapidly with temperatures higher than 500 deg C. Alloy additions of Cr, Si, titanium, and molybdenum, alone or in combination, show resistance up to 600 deg C. Where applicable, the attack of ferrous alloys by mercury can be reduced to negligible amounts by the addition of 10 ppm Ti to the mercury. This raises the useful range of operating temperatures to 650 deg C. Additions of metal with a higher affinity for O2 than titanium, such as sodium or magnesium, may be needed to prevent oxidation of the titanium and loss of the inhibitive action.
- Aluminum – Plain C steels are not satisfactory for long term containment of molten aluminum.
- Gallium – Gallium is one of the most aggressive of all liquid metals and cannot be contained by C steels or low alloy steels at high temperatures.
- Indium – Plain C steels and low alloy steels have poor resistance to molten indium.
- Lead, bismuth, tin, and their alloys – Low alloy steels have good resistance to lead up to 600 deg C, to bismuth up to 700 deg C, and to tin only up to 150 deg C. The various alloys of lead, bismuth, and tin are more aggressive.

Leave a Comment